
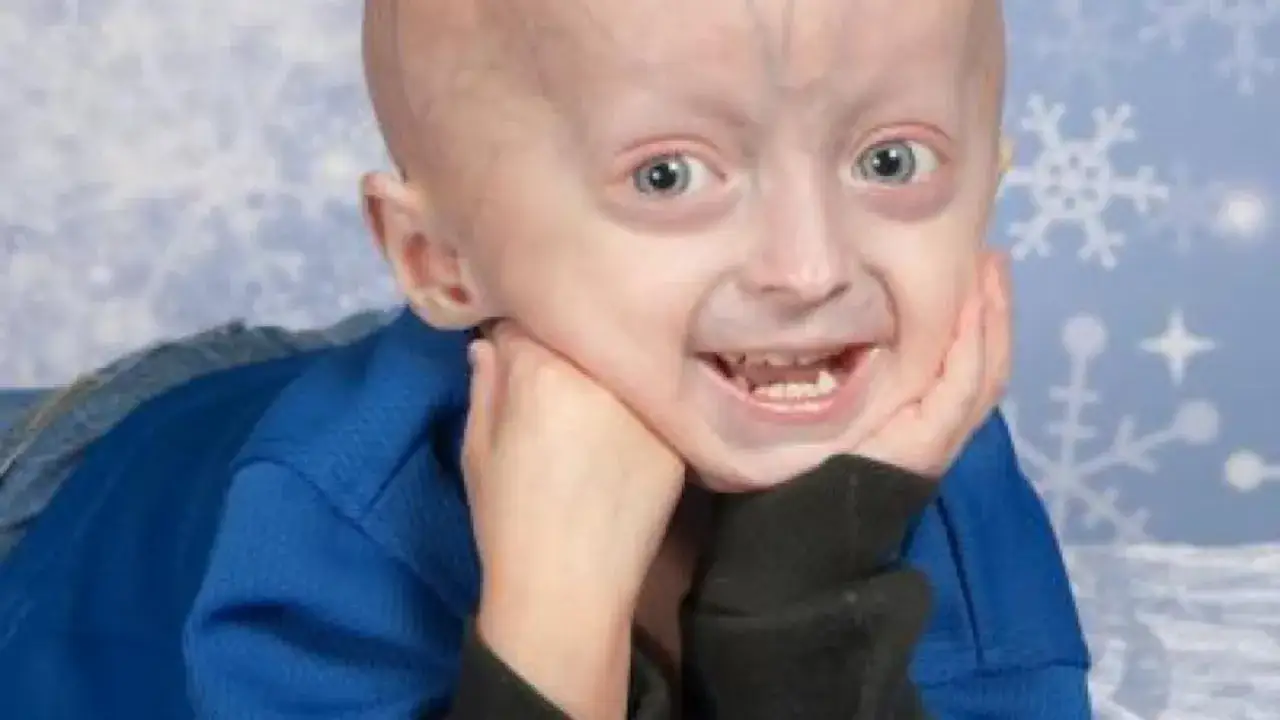
Progeria zwykle kojarzy się z dzieckiem wyglądającym na osobę starszą, ale ten obraz przesłania najważniejszy mechanizm choroby. Chodzi o ultrarzadki zespół genetyczny, w którym wada białek otoczki jądra komórkowego uruchamia przedwczesne starzenie tkanek, zwłaszcza naczyń krwionośnych. Największe ryzyko nie dotyczy wyglądu, lecz serca, naczyń, wzrostu i codziennego bezpieczeństwa dziecka. W praktyce liczy się szybkie rozpoznanie, dobre różnicowanie i opieka wielospecjalistyczna.
Progeria łączy wczesny początek objawów z dominującym ryzykiem sercowo-naczyniowym
- Klasyczna HGPS najczęściej wynika z mutacji LMNA pojawiającej się de novo i zwykle ujawnia się w pierwszych dwóch latach życia.
- Rozwój intelektualny bywa prawidłowy, choć wzrost, tkanka tłuszczowa, skóra, stawy i zęby rozwijają się nietypowo.
- Lonafarnib jest obecnie pierwszą terapią ukierunkowaną dopuszczoną dla części pacjentów z HGPS, ale nie zastępuje stałego nadzoru specjalistycznego.
- W Polsce zespół progerii Hutchinsona i Gilforda figuruje w portalu chorób rzadkich pod kodem ORPHA 740 i ICD-10 E34.8.
Czym jest progeria i dlaczego nie chodzi tylko o starzenie się skóry?
Progeria to rzadki zespół genetyczny, a nie zwykłe „szybsze starzenie”. W klasycznej postaci, czyli zespole progerii Hutchinsona i Gilforda, problem zaczyna się od nieprawidłowego przetwarzania laminy A, co prowadzi do powstawania progeryny i uszkodzenia komórkowej struktury podtrzymującej jądro. Według Progeria Research Foundation obecnie żyje 163 dzieci i młodych dorosłych z HGPS oraz 87 osób z progeroidową laminopatią w 55 krajach, co dobrze pokazuje skalę tej ultrarzadkości.
To, co z zewnątrz wygląda jak przyspieszone starzenie, w środku oznacza przede wszystkim chorobę naczyń i zaburzenie wzrastania. Dziecko może wyglądać na zdrowe po urodzeniu, a pierwsze sygnały pojawiają się później: słabszy przyrost masy, utrata tkanki tłuszczowej, łysienie, sztywność stawów i charakterystyczne rysy twarzy. W klasycznym HGPS rozwój umysłowy pozostaje zazwyczaj prawidłowy, więc choroba łatwo bywa mylona z problemem dermatologicznym, ortopedycznym albo „po prostu drobną budową ciała”.
Najtrudniejszy do przeoczenia, a jednocześnie najważniejszy prognostycznie, jest układ sercowo-naczyniowy. U nieleczonych osób średnia długość życia wynosi około 14,5 roku, a przy leczeniu lonafarnibem wydłuża się przeciętnie do około 18,7 roku. Różnica nie oznacza wyleczenia, ale pokazuje, że choroba nie jest już wyłącznie diagnozą bez realnych narzędzi terapeutycznych.
Zapamiętaj: progeria nie jest jednym objawem i nie sprowadza się do „starego wyglądu”. Największe znaczenie ma ryzyko miażdżycy, udaru i niewydolności serca.
Przeczytaj również: Neurofibromatoza typu 1 - Jak rozpoznać i kontrolować objawy NF1?
Jak rozpoznać progerię i odróżnić ją od innych zespołów progeroidowych?
Rozpoznanie opiera się na obrazie klinicznym, ale potwierdza je badanie molekularne LMNA. W GeneReviews podkreślono, że HGPS należy podejrzewać przy prawidłowym rozwoju intelektualnym, zahamowaniu wzrostu, lipodystrofii, łysieniu, zmianach skórnych i cechach kostnych, które pojawiają się zwykle wcześnie. To ważne, bo zewnętrzny obraz może być sugestywny, ale nie wystarcza do pewnego rozpoznania.
Pierwsze objawy
Pierwsze sygnały zwykle nie są dramatyczne, tylko powolne i bardzo niespecyficzne. Rodzice często zauważają, że dziecko rośnie wolniej niż rówieśnicy, słabiej przybiera na wadze, ma mniej tkanki tłuszczowej pod skórą, a twarz staje się bardziej „wyostrzona”: duża głowa względem twarzy, wąski nos, mała żuchwa i cienkie wargi. Do tego dochodzą sztywność stawów, łysienie, zmiany paznokci, wysoki głos, problemy stomatologiczne oraz skóra o twardawym, napiętym wyglądzie.
Wczesne objawy bywają mylące, bo oddzielnie pasują do różnych specjalności. Dziecko może trafić do dermatologa z powodu skóry, do ortopedy z powodu bioder lub stawów, do stomatologa z powodu opóźnionego wyrzynania zębów albo do pediatry z powodu słabego przyrostu masy. Rozpoznanie staje się łatwiejsze dopiero wtedy, gdy objawy zaczynają układać się w jeden spójny wzór.
Jak potwierdza się rozpoznanie
W klasycznym HGPS najczęściej chodzi o wariant de novo, czyli nową zmianę genetyczną pojawiającą się po raz pierwszy u dziecka. GeneReviews podaje, że około 98% przypadków wynika z takiej zmiany, a około 2% wiąże się z mozaicyzmem gonadalnym jednego z rodziców, więc ryzyko powtórzenia w rodzinie jest niskie, ale niezerowe. To ma znaczenie dla poradnictwa genetycznego, planowania kolejnej ciąży i wyjaśnienia, dlaczego rodzice zazwyczaj nie mają własnych objawów.
Badanie genetyczne może wykazać klasyczny wariant LMNA c.1824C>T albo inną zmianę prowadzącą do powstawania progeryny. Gdy obraz kliniczny jest niejednoznaczny, pomocne bywa szersze badanie, na przykład panel genów, eksom albo genom, zwłaszcza jeśli choroba przypomina inne zespoły progeroidowe. W praktyce liczy się nie tylko nazwa mutacji, ale też to, czy zmiana rzeczywiście tłumaczy cały obraz choroby.
Uwaga: pojedynczy prawidłowy wynik nie zamyka sprawy, jeśli objawy dopiero się rozwijają. W progerii obraz kliniczny potrafi dojrzewać stopniowo.
Z czym najczęściej się myli
| Wariant | Początek | Najważniejsze cechy | Co odróżnia od klasycznej progerii |
|---|---|---|---|
| Klasyczna HGPS | W pierwszych latach życia | Spowolniony wzrost, lipodystrofia, alopecja, sztywność stawów, wczesna miażdżyca | Najczęściej progerina i wariant LMNA związany z HGPS |
| Nieklasyczna HGPS | Dzieciństwo, czasem później | Podobne cechy, ale łagodniejsze albo mniej typowe | Inny wariant LMNA, przebieg i tempo zmian mogą być mniej oczywiste |
| Zespół Wernera | Po okresie dojrzewania | Przedwczesne starzenie, zaćma, cukrzyca, owrzodzenia, nowotwory | Późniejszy początek i inny gen, zwykle WRN |
| Wiedemann-Rautenstrauch / Nestor-Guillermo | Przed urodzeniem lub w niemowlęctwie | Niska masa urodzeniowa, lipodystrofia, cechy progeroidowe, czasem osteoliza | Inny przebieg, inna genetyka i często odmienny profil powikłań |
Różnicowanie ma znaczenie, bo nie każda choroba „podobna do progerii” daje ten sam profil powikłań i ten sam kierunek leczenia. W HGPS największy ciężar spoczywa na naczyniach, sercu i kościach, ale w innych zespołach progeroidowych może dominować inny układ, na przykład neurologiczny, metaboliczny albo onkologiczny. Sama sylwetka dziecka nigdy nie powinna być jedyną podstawą wnioskowania.
Jak wygląda leczenie i codzienny nadzór medyczny?
Leczenie nie usuwa przyczyny, ale spowalnia część powikłań i porządkuje opiekę wielospecjalistyczną. W HGPS standardem pozostaje połączenie terapii ukierunkowanej, monitorowania serca i naczyń, rehabilitacji, wsparcia żywieniowego, opieki stomatologicznej oraz kontroli wzroku i słuchu. To jedna z tych chorób, w których dobrze zaplanowana rutyna ma większe znaczenie niż pojedyncza spektakularna interwencja.
Leczenie ukierunkowane
Najważniejszą zmianą jest lonafarnib, czyli inhibitor farnesylotransferazy, który zmniejsza skutki powstawania progeryny. Według FDA lek jest zatwierdzony dla pacjentów od 12. miesiąca życia przy odpowiedniej powierzchni ciała, a EMA dopuściła go w Unii Europejskiej dla dzieci z genetycznie potwierdzoną HGPS oraz niektórymi progeroidowymi laminopatiami. To nie jest leczenie przyczynowe w sensie pełnego odwrócenia choroby, ale realnie zmienia przebieg wielu przypadków.
W badaniach i obserwacjach klinicznych lonafarnib poprawiał sztywność naczyń, część parametrów krążenia, słuch w zakresie niskich częstotliwości oraz częstotliwość bólów głowy. W praktyce oznacza to, że dziecko nadal wymaga stałej opieki, ale pewne powikłania mogą rozwijać się wolniej. Warto też pamiętać o działaniach niepożądanych, głównie żołądkowo-jelitowych, bo to one najczęściej wpływają na tolerancję terapii.
Nadzór i codzienna opieka
Codzienność opiera się na nadzorze wielospecjalistycznym. Według GeneReviews po rozpoznaniu zaleca się ocenę wzrostu i odżywienia, badanie stomatologiczne, ocenę skóry, konsultację ortopedyczną, EKG, echokardiografię, badanie tętnic szyjnych, ocenę neurologiczną, okulistyczną i audiologiczną oraz stałe poradnictwo rodzinne. Część aktywności ma charakter rutynowy, a część jest reakcją na wczesne sygnały pogarszania się układu naczyniowego.
| Obszar | Częstotliwość | Po co to się robi |
|---|---|---|
| Wzrost i odżywienie | Przy każdej wizycie | Żeby wychwycić spowolnienie przyrostu masy i ocenę podaży kalorii |
| Stomatologia | Co 12 miesięcy | Żeby kontrolować wyrzynanie zębów, stłoczenie i próchnicę |
| Układ sercowo-naczyniowy | Co 6-12 miesięcy | Żeby śledzić miażdżycę, ciśnienie, echo i przepływy w naczyniach |
| Neurologia | Co 6-12 miesięcy | Żeby wychwycić bóle głowy, incydenty niedokrwienne i objawy udaru |
| Wzrok i słuch | Co 12 miesięcy | Żeby kontrolować suche oczy, uszkodzenie rogówki i niedosłuch |
W praktyce: ciężka miażdżyca może rozwijać się zanim EKG pokaże duże odchylenia. Dlatego przy progerii nie wystarcza samo „brak objawów”, potrzebny jest regularny nadzór naczyń i serca.
Istnieją też praktyczne ograniczenia, które łatwo przeoczyć. Znieczulenie ogólne i intubacja wymagają szczególnej ostrożności, bo u osób z HGPS anatomia dróg oddechowych i sztywność naczyń zwiększają ryzyko powikłań. Niektóre dzieci źle tolerują odwodnienie, gorączkę, anemię i zbyt intensywną aktywność, a suplementacja wapnia nie jest dobrym odruchem „na wszelki wypadek”, jeśli może nasilać zwapnienia pozaszkieletowe.
Przeczytaj również: Choroby rzadkie - Jak rozpoznać objawy i gdzie szukać pomocy?
Jakie są realia w Polsce i gdzie zaczyna się droga pacjenta?
W Polsce najpraktyczniejszym punktem startu jest genetyk kliniczny i portal chorób rzadkich. W portalu Choroby Rzadkie HGPS widnieje pod kodem ORPHA 740, OMIM 176670, ICD-10 E34.8 i ICD-11 LD2B, a obok opisu dostępne są odsyłacze do ośrodków eksperckich, poradni, programów lekowych NFZ, badań klinicznych, hospicjów i rehabilitacji. To ważne, bo w chorobach ultrarzadkich sama diagnoza bez systemu prowadzenia bywa niewystarczająca.
Najbezpieczniejsza ścieżka wygląda zwykle tak: najpierw ocena pediatryczna lub internistyczna, potem skierowanie do genetyka klinicznego, badanie molekularne, a równolegle baseline kardiologiczny, ortopedyczny i okulistyczny. Jeśli potwierdzi się HGPS, dobry plan obejmuje poradnictwo genetyczne, ustalenie harmonogramu kontroli i wskazanie ośrodka, który widzi takie dzieci na co dzień. W chorobie tak rzadkiej każdy miesiąc chaosu diagnostycznego oznacza utratę czasu, którego nie da się odzyskać.
Najczęstszy błąd polega na założeniu, że dziecko jest po prostu bardzo drobne albo „dziwnie wygląda” i że problem sam się wyjaśni. Drugi błąd to mylenie progerii z innymi zespołami progeroidowymi lub z rozproszonymi problemami dermatologicznymi i ortopedycznymi, przez co rozpoznanie rozciąga się na wiele wizyt. Trzeci błąd to opieranie się wyłącznie na badaniach krwi, podczas gdy w HGPS lipidogram bywa zaskakująco mało charakterystyczny, a ryzyko naczyniowe i tak pozostaje wysokie.
Kiedy trzeba reagować pilnie
Wystąpienie nagłego bólu w klatce piersiowej, asymetrii twarzy, zaburzeń mowy, osłabienia kończyny, omdlenia, silnego bólu głowy albo duszności wymaga pilnej oceny medycznej. U dziecka z progerią takie objawy mogą oznaczać zdarzenie naczyniowe, a nie zwykłe osłabienie czy przejściowy spadek formy. Zbyt długie czekanie bywa w tej chorobie bardziej ryzykowne niż sam wynik diagnostyczny.
W rodzinach, w których pojawia się pytanie o kolejne dzieci, potrzebne jest też spokojne poradnictwo genetyczne. Ryzyko nawrotu jest niskie, ale niezerowe z powodu mozaicyzmu, więc decyzje reprodukcyjne powinny opierać się na konkretnym wyniku genetycznym, a nie na przypuszczeniach. To właśnie dlatego w progerii tak ważne jest połączenie diagnostyki molekularnej z rozmową o rodzinie, a nie tylko z opisem objawów.
W praktyce: najlepszy efekt daje prowadzenie dziecka w zespole, który łączy genetykę, kardiologię, ortopedię, okulistykę i rehabilitację. W tej chorobie dobra koordynacja jest równie ważna jak sam lek.
Najbezpieczniej prowadzić progerię w ośrodku, który łączy genetykę, kardiologię, rehabilitację i regularny nadzór nad powikłaniami naczyń.