Wiele osób kojarzy dystrofię mięśniową Duchenne'a głównie z trudniejszym chodzeniem, choć choroba szybko wykracza poza same mięśnie kończyn. W praktyce chodzi o brak dystrofiny, przez co stopniowo słabną mięśnie szkieletowe, oddechowe i serce. Ten tekst porządkuje objawy, diagnostykę, leczenie i realne ograniczenia dostępne obecnie w Polsce.
DMD ujawnia się wcześnie i wymaga potwierdzenia genetycznego
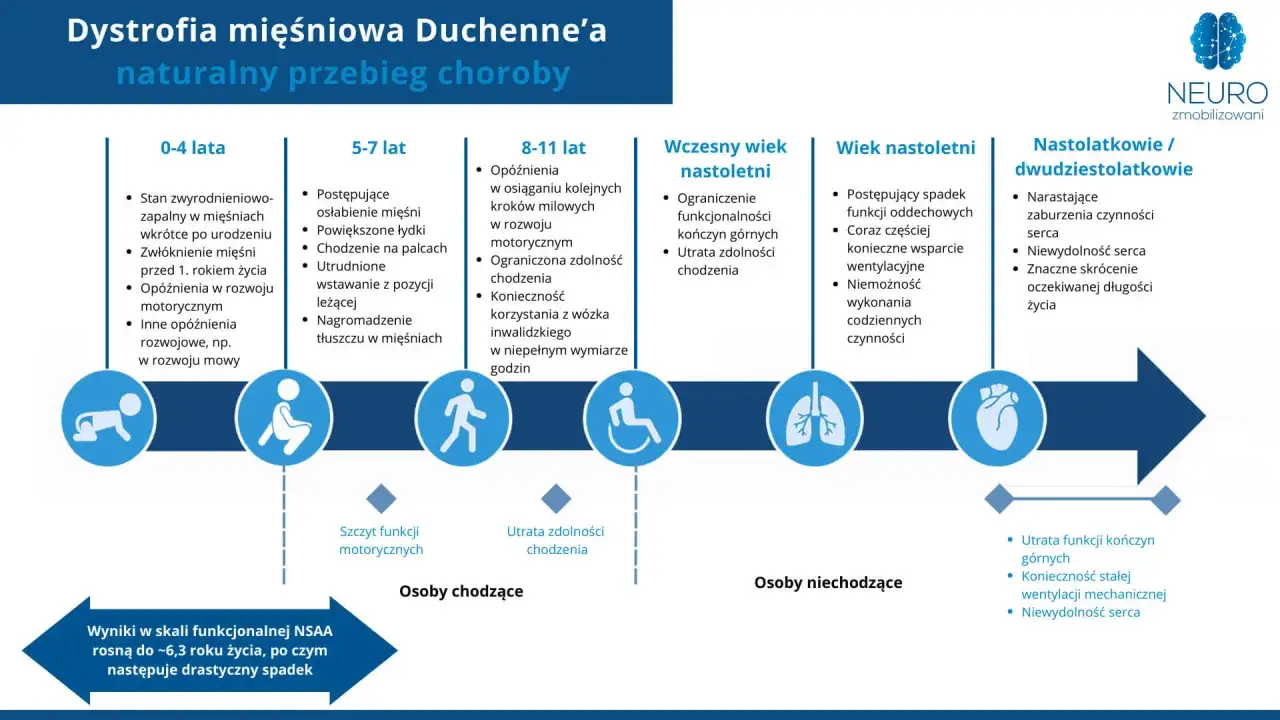
- DMD najczęściej ujawnia się około 3.-4. roku życia, gdy pojawiają się trudności z wstawaniem z podłogi i chodzeniem na palcach.
- Kinaza kreatynowa CK bywa podwyższona 10-100 razy, co odróżnia tę chorobę od zwykłego zmęczenia mięśni.
- Badanie MLPA wykrywa około 70% mutacji prowadzących do DMD, a sekwencjonowanie dopełnia diagnostykę większości pozostałych przypadków.
- Kortykosteroidy mogą wydłużyć czas samodzielnego chodzenia o 1,5-2 lata, jeśli leczenie jest prowadzone pod kontrolą specjalistów.
- Agamree i Duvyzat są dziś zarejestrowane w UE, ale kwalifikacja i dostęp nadal zależą od kraju oraz programu leczenia.
Dlaczego dystrofia mięśniowa Duchenne'a szybko przestaje być tylko problemem mięśni?
To choroba dystrofiny, a nie samej kondycji, dlatego obejmuje cały układ ruchu, oddech i serce. Według oficjalnego opisu choroby w serwisie Choroby Rzadkie DMD jest rzadką chorobą genetyczną sprzężoną z chromosomem X, a częstość urodzeń chłopców wynosi około 1 na 3500. Gdy dystrofina zanika, włókna mięśniowe stają się podatne na mikrouszkodzenia, stan zapalny i bliznowacenie, dlatego z czasem w grę wchodzi nie tylko chodzenie, ale też kardiomiopatia i niewydolność oddechowa.
Mutacja, która niszczy dystrofinę
W DMD uszkodzony jest gen DMD na chromosomie X, a dziedziczenie ma zwykle charakter recesywny. To właśnie dlatego choroba typowo dotyczy chłopców, a kobiety najczęściej pozostają nosicielkami, choć objawowa postać może pojawić się także u dziewcząt. Najczęstsze mutacje to delecje, rzadziej duplikacje i mutacje punktowe, ale efekt biologiczny jest ten sam: organizm nie produkuje prawidłowej dystrofiny albo produkuje jej zbyt mało.
Pierwsze objawy, które łatwo przeoczyć
Najwcześniej widać opóźnienie rozwoju ruchowego, chód na palcach, przerost łydek i trudność z wstawaniem z podłogi. Charakterystyczny jest objaw Gowersa, czyli „wspinanie się po sobie” przy podnoszeniu z pozycji leżącej lub kucającej. U części dzieci pojawia się też opóźnienie mowy albo łagodne trudności poznawcze, więc obraz bywa mylący i zbyt łatwo przypisywany „słabszej sprawności” albo indywidualnemu tempu rozwoju.
Zapamiętaj: DMD nie zaczyna się od nagłego paraliżu. Najpierw pojawia się subtelna utrata sprawności, a dopiero później zajęcie oddechu i serca.
Dlaczego płeć nie wyczerpuje tematu
Dziedziczenie sprzężone z chromosomem X nie zamyka sprawy na prostym podziale „chłopcy chorują, dziewczynki nie”. W praktyce kobiety najczęściej są nosicielkami, ale mogą przekazywać mutację dalej, a incydentalnie występują też pełnoobjawowe postacie u kobiet. To jeden z powodów, dla których wywiad rodzinny, diagnostyka genetyczna i nieupraszczanie obrazu klinicznego są ważniejsze niż sama obserwacja „czy dziecko jeszcze chodzi”.
Przeczytaj również: Rdzeniowy zanik mięśni (SMA) - Jak rozpoznać objawy i kiedy reagować

Jak rozpoznaje się dystrofię mięśniową Duchenne'a bez zbędnych badań?
Rozpoznanie zaczyna się od prostych badań, ale kończy na teście genetycznym, bo sam obraz kliniczny bywa mylący. Najpierw szuka się cech uszkodzenia mięśni, potem potwierdza konkretną mutację, bo to właśnie ona decyduje o rozpoznaniu, poradnictwie rodzinnym i kwalifikacji do leczenia. Im szybciej to się stanie, tym mniej miejsca zostaje na niepotrzebne badania i opóźnienia.
CK i enzymy mięśniowe
W pierwszym etapie diagnostyki zwykle widać bardzo wysoką aktywność CK, często od 10 do 100 razy powyżej normy. Równocześnie mogą rosnąć także aminotransferazy ALT i AST, co bywa mylące, bo nie oznacza automatycznie choroby wątroby. W DMD to efekt rozpadu włókien mięśniowych, a nie izolowanego problemu hepatologicznego.
Test genetyczny
Badanie MLPA wykrywa delecje i duplikacje odpowiadające za około 70% mutacji prowadzących do DMD, a później wykonuje się sekwencjonowanie genu, które obejmuje kolejne około 28-30% przypadków. Brak mutacji w MLPA nie wyklucza choroby. W praktyce oznacza to, że ujemny wynik jednego etapu nie zamyka diagnostyki, tylko uruchamia następny.
Uwaga: Ujemny wynik MLPA nie kończy sprawy. Przy mocnym podejrzeniu DMD trzeba iść dalej, bo część mutacji wychodzi dopiero w sekwencjonowaniu.
Czego zwykle nie trzeba robić
Badanie EMG i biopsja mięśnia zwykle nie są konieczne, jeśli obraz kliniczny i genetyka są wystarczająco jednoznaczne. To ważne, bo wiele rodzin trafia do diagnostyki z nastawieniem na kolejne, bardziej inwazyjne kroki, podczas gdy właściwy trop daje już genetyka. Potwierdzenie mutacji nie służy wyłącznie nazwaniu choroby, ale też ustaleniu, czy w grę wchodzą terapie zależne od konkretnego wariantu genu i jak prowadzić badania u krewnych.
Przeczytaj również: Choroba Hawkinga - Czym jest ALS i dlaczego przebieg bywa nietypowy
Jakie leczenie jest dziś realne i co działa najlepiej?
Najlepiej działa połączenie leczenia objawowego, rehabilitacji, ochrony serca i płuc oraz leków dobranych do wieku i typu mutacji. W DMD nie ma jednej terapii, która „załatwia sprawę” sama, dlatego największą różnicę robi zespół specjalistów, a nie pojedyncza recepta. Właśnie tu najlepiej widać, że szybkie rozpoznanie przekłada się na realny zysk funkcjonalny.
Kortykosteroidy jako baza
W oficjalnym opisie choroby zalecane jest rozpoczęcie doustnych kortykosteroidów około 4. roku życia, najczęściej w postaci prednizonu 0,75 mg/kg/dobę albo deflazakortu 0,9 mg/kg/dobę, z maksymalnymi dawkami odpowiednio 30 mg i 36 mg na dobę. Taka terapia może wydłużyć czas samodzielnego chodzenia o 1,5-2 lata, zmniejszyć ryzyko skoliozy i opóźnić rozwój przewlekłej niewydolności oddechowej. To nie jest leczenie pozbawione kosztów dla organizmu, ale w DMD nadal pozostaje filarem, od którego zaczyna się większość planów terapeutycznych.
W praktyce: Sterydów nie dobiera się „na oko”. Liczą się wiek, funkcja ruchowa, tolerancja działań niepożądanych i plan szczepień przed startem leczenia.
Nowe leki w Europie
W UE pojawiły się już leki, które adresują inne punkty przebiegu choroby niż klasyczne sterydy. Agamree jest przeznaczony dla pacjentów od 2. roku życia i podawany doustnie raz dziennie, a Duvyzat dotyczy chodzących pacjentów od 6. roku życia, którzy już otrzymują kortykosteroidy. Oba leki wymagają prowadzenia przez lekarza doświadczonego w DMD, bo nie chodzi tylko o samo przepisanie preparatu, ale też o kontrolę działań niepożądanych i ocenę odpowiedzi na leczenie.
| Opcja | Kto zwykle się kwalifikuje | Jak podaje się lek | Najważniejsze ograniczenie |
|---|---|---|---|
| Prednizon | Dzieci z DMD, zwykle od około 4. roku życia | 0,75 mg/kg/dobę, maksymalnie 30 mg | Wymaga stałej kontroli wzrostu, masy ciała, kości i tolerancji |
| Deflazakort | Dzieci z DMD, zwykle od około 4. roku życia | 0,9 mg/kg/dobę, maksymalnie 36 mg | Podobny cel jak prednizon, ale profil tolerancji może być inny |
| Agamree | Pacjenci od 2. roku życia | Zawiesina doustna raz dziennie | Wymaga prowadzenia przez specjalistę i obserwacji działań steroidopodobnych |
| Duvyzat | Chodzący pacjenci od 6. roku życia na kortykosteroidach | Płyn doustny dwa razy dziennie | Trzeba monitorować płytki krwi, triglicerydy i biegunkę |
W Polsce takie decyzje nie przekładają się automatycznie na dostęp przy łóżku pacjenta. W analizie AOTMiT dotyczącej giwinostatu podkreślono, że dostęp refundacyjny w DMD pozostaje bardzo ograniczony, więc kluczowa bywa kwalifikacja do właściwego programu i ośrodka prowadzącego. To właśnie różni teorię od praktyki: lek może być zarejestrowany, ale w konkretnym kraju nadal wymaga przejścia przez własną ścieżkę finansowania.
Serce, płuca i kości
W DMD nie wystarczy patrzeć na nogi, bo choroba wymaga równoległej kontroli kardiologicznej i oddechowej. Oficjalne zalecenia mówią o regularnej ocenie EKG, Holtera i echo serca, a także o ocenie funkcji płuc, szczególnie w drugiej dekadzie życia i przed znieczuleniem ogólnym. Gdy pojawiają się objawy przewlekłej niewydolności oddechowej, potrzebne bywa nieinwazyjne wsparcie oddechowe, a przy leczeniu sterydami trzeba też myśleć o szczepieniach i zdrowiu kości.
Polskie protokoły NFZ porządkują leczenie chorych na DMD w modelu koordynowanym, więc warto szukać prowadzenia w takim schemacie zamiast rozbijania opieki na pojedyncze wizyty. NFZ publikuje protokoły dla świadczeniodawców, a to dla pacjenta oznacza jedno: najlepiej działa opieka, w której neurolog, kardiolog, pulmonolog, rehabilitant, dietetyk i genetyk pracują według jednego planu.
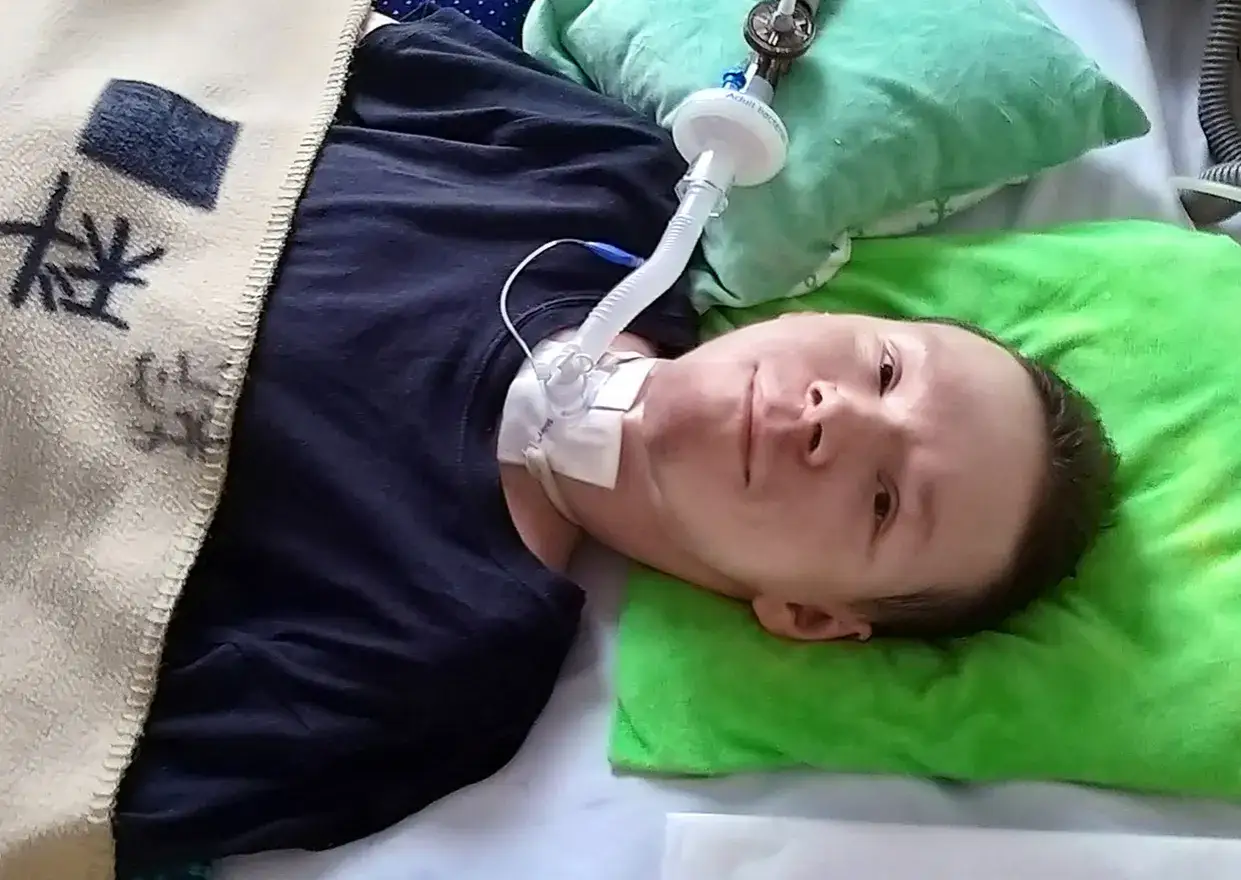
Czym DMD różni się od dystrofii Beckera i kiedy pojawiają się pomyłki?
Becker jest zwykle wolniejsza, bo dystrofina nie znika całkiem, ale obraz bywa podobny na tyle, że rozpoznanie potrafi się opóźnić. W DMD białko jest praktycznie nieobecne, a w dystrofii Beckera jego ilość lub jakość jest częściowo zachowana, dlatego przebieg jest łagodniejszy i bardziej rozciągnięty w czasie. To rozróżnienie jest ważne nie tylko akademicko, lecz także praktycznie, bo determinuje tempo utraty sprawności i sposób prowadzenia rodziny.
Różnica w dystrofinie
Najkrócej można to ująć tak: Duchenne oznacza prawie całkowity brak dystrofiny, a Becker jej częściowe zachowanie. W praktyce DMD zaczyna się wcześniej, szybciej odbiera samodzielny chód i wcześniej zajmuje serce oraz oddech. Przy Beckerze dziecko lub nastolatek może przez dłuższy czas funkcjonować „w miarę normalnie”, co potrafi uśpić czujność.
Najczęstsze pomyłki
Jednym z częstszych błędów jest uznanie dziecka za mniej sprawne ruchowo, ale zdrowe, i odsyłanie do rehabilitacji bez pełnej diagnostyki. Inny klasyczny trop to niepotrzebne skupienie się na podwyższonych ALT i AST, jakby źródłem problemu była wątroba, podczas gdy mięśnie już się rozpadają. Gdy w tle jest opóźnienie ruchowe, chód na palcach i objaw Gowersa, same obserwacje ortopedyczne zwykle nie wystarczają.
Sytuacje graniczne
Najbardziej zdradliwe są przypadki, w których objawy rozwijają się wolniej albo obraz nie pasuje do „podręcznikowego chłopca z DMD”. Dotyczy to między innymi dziewcząt z objawowym nosicielstwem, dzieci z łagodniejszym początkiem, a także rodzin, w których wcześniej nikt nie miał postawionego rozpoznania. W takich sytuacjach wynik genetyczny jest nie tylko potwierdzeniem diagnozy, ale też drogą do poradnictwa rodzinnego i rozważenia terapii ukierunkowanych na konkretny wariant mutacji.
Zapamiętaj: Gdy dziecko ma stale podwyższoną CK, objaw Gowersa i dodatni wywiad rodzinny, czekanie na „spontaniczną poprawę” zwykle tylko opóźnia leczenie.
Jak wygląda ścieżka działania, gdy pojawia się podejrzenie DMD?
Najpierw trzeba dotrzeć do neurologa dziecięcego albo ośrodka chorób rzadkich, a potem szybko potwierdzić mutację i ustawić opiekę. W DMD nie działa strategia „zobaczymy za kilka miesięcy”, bo choroba postępuje i zabiera sprawność zanim rodzina zdąży się oswoić z samym podejrzeniem. Im wcześniej powstanie jeden plan, tym lepiej dla ruchu, serca i oddechu.
Pierwszy kontakt
W praktyce rozsądny pierwszy krok to pediatra lub lekarz rodzinny, który widzi regres sprawności, a następnie skierowanie do neurologa, genetyka i ośrodka wyspecjalizowanego w chorobach rzadkich. To właśnie tam najłatwiej zorganizować test genetyczny, interpretację wyniku i ocenę dalszych potrzeb. Warto od razu myśleć o poradnictwie rodzinnym, bo choroba dziecka zmienia także ocenę ryzyka u rodzeństwa i krewnych.
- Zgłoś objawy takie jak chód na palcach, objaw Gowersa, regres biegania albo trudność z wchodzeniem po schodach.
- Poproś o CK i skierowanie do testu genetycznego zamiast czekać na kolejne niespójne konsultacje.
- Ustal poradnię genetyczną, bo wynik zmienia także ocenę ryzyka w rodzinie.
- Zapewnij bazową ocenę serca i płuc, zanim pojawi się utrata chodu albo nasilona duszność.
Co ustawić od razu
Już na starcie potrzebne są rehabilitacja, plan kontroli kardiologicznej, ocena oddechu i ustalenie żywienia, a nie tylko sama nazwa rozpoznania. Przy przewlekłej steroidoterapii trzeba też osobno omówić szczepienia, bo oficjalny opis choroby zwraca uwagę, że szczepionek żywych nie łączy się z leczeniem bez wcześniejszego uzgodnienia z lekarzem. To ważny detal, bo wielu rodziców myśli o sterydach wyłącznie jako o „leku na mięśnie”, a to terapia wpływająca na cały organizm.
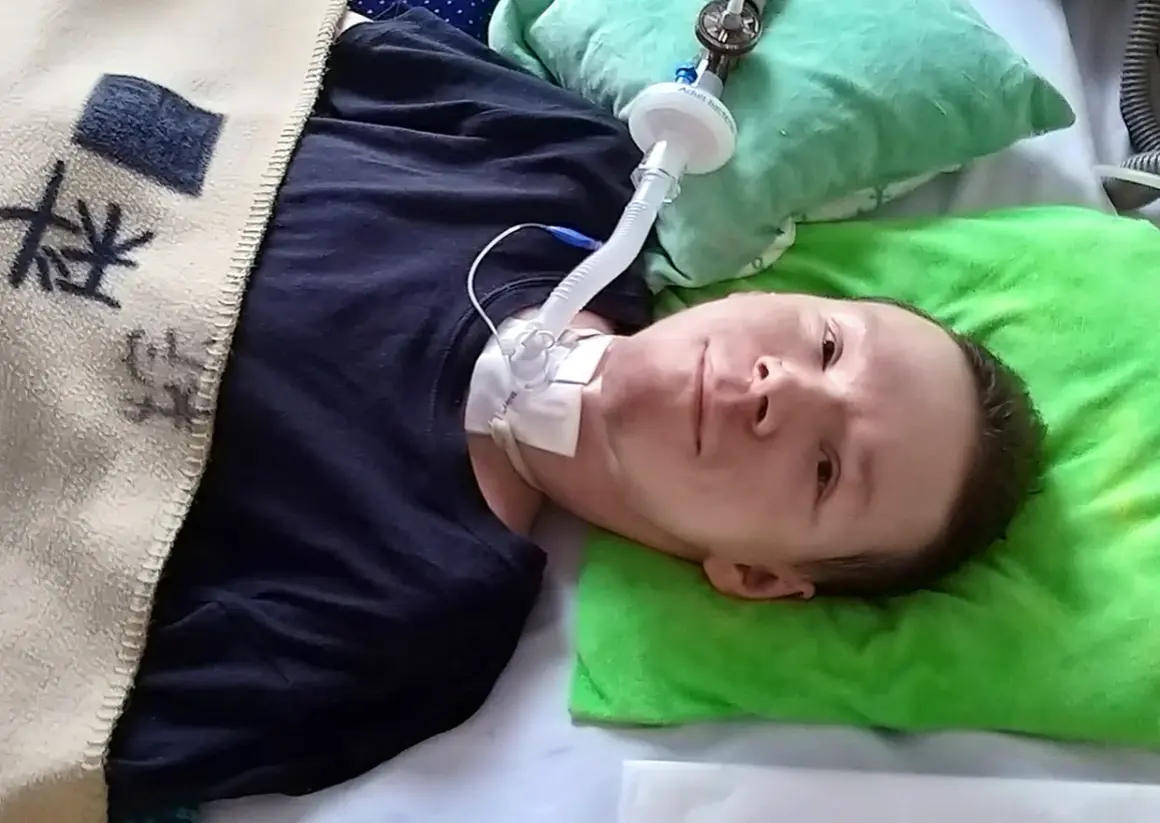
Kiedy reagować pilnie
Pilnego kontaktu z lekarzem wymagają narastająca duszność, częste infekcje oddechowe, problemy z połykaniem, wybudzanie się w nocy z powodu oddechu i wyraźny spadek tolerancji wysiłku. To właśnie wtedy wchodzi do gry ośrodek wentylacji domowej, a nie tylko rehabilitacja. Jeśli dziecko zaczyna męczyć się przy jedzeniu albo ma bezdechy w nocy, nie warto czekać na kolejny „spokojniejszy tydzień”.
W praktyce: Najlepszy plan przy DMD to jeden ośrodek prowadzący, jeden harmonogram kontroli i szybkie decyzje o oddechu, sercu oraz rehabilitacji. Rozproszenie opieki zwykle kosztuje funkcję ruchową.
W DMD najwięcej zmienia szybkie potwierdzenie mutacji, opieka wielospecjalistyczna i regularne monitorowanie oddechu, serca oraz sprawności ruchowej.