ALD potrafi przez lata wyglądać jak problem neurologiczny, hormonalny albo szkolny, zanim ktoś połączy wszystkie tropy. Właśnie dlatego adrenoleukodystrofia bywa rozpoznawana dopiero wtedy, gdy część uszkodzeń jest już zaawansowana. W tekście poniżej pojawia się zarówno obraz choroby, jak i praktyczna ścieżka postępowania w Polsce.
ALD łączy neurologię i endokrynologię
- ABCD1 to gen związany z ALD, a jego uszkodzenie prowadzi do gromadzenia bardzo długich kwasów tłuszczowych i uszkodzeń tkanek.
- U chłopców i mężczyzn choroba częściej daje cięższy przebieg, ale kobiety także mogą mieć objawy w dorosłości.
- Monitorowanie ACTH i kortyzolu co 3–6 miesięcy u młodszych chłopców ogranicza ryzyko przeoczenia niewydolności nadnerczy.
- MRI co 6 miesięcy w wieku 3–12 lat pomaga wykryć mózgową postać zanim pojawią się wyraźne objawy kliniczne.
- W Polsce obowiązujący panel przesiewu noworodków obejmuje inne choroby, więc ALD wymaga czujności klinicznej i rodzinnej.
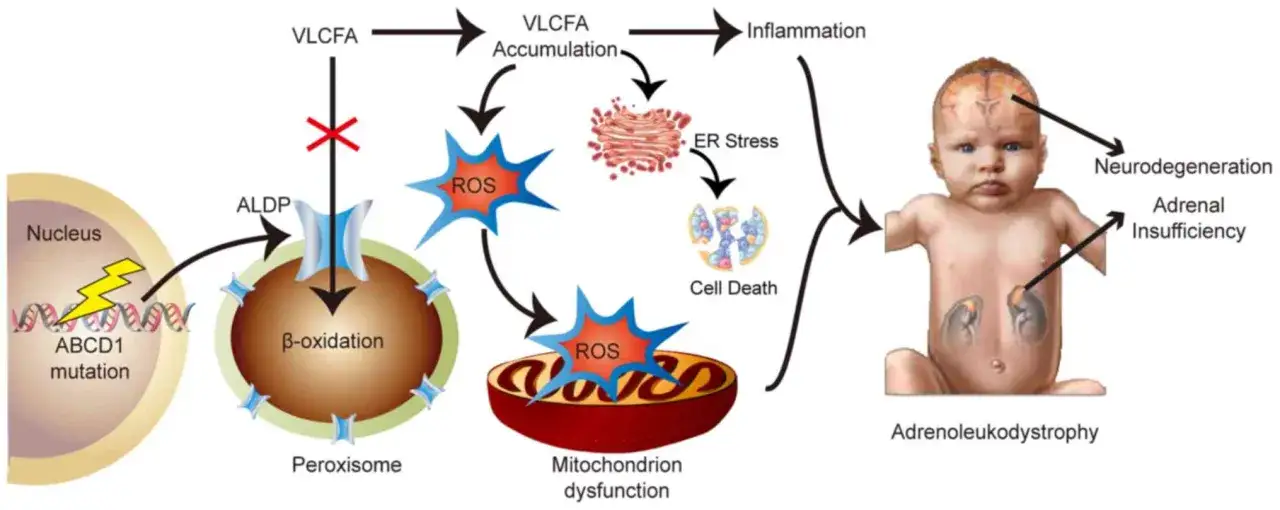
Czym jest ALD i dlaczego łatwo je przeoczyć?
ALD to rzadka, sprzężona z chromosomem X choroba peroksysomalna, która uszkadza nadnercza i białą materię mózgu. Objawy zależą od wieku, płci i tego, która część układu nerwowego lub hormonalnego została zajęta jako pierwsza. Polski portal Choroby Rzadkie opisuje ją jako postępujące zaburzenie z niewydolnością nadnerczy, mielopatią i neuropatią obwodową.
Najbardziej mylące jest to, że ALD nie ma jednego, stałego obrazu. U części osób zaczyna się od problemów z zachowaniem, u innych od sztywności nóg, a jeszcze u innych od niewydolności kory nadnerczy. Ta zmienność sprawia, że choroba potrafi naśladować kilka różnych specjalności jednocześnie, dlatego sam „dziwny” zestaw objawów bywa cenniejszy niż pojedyncza dolegliwość.
Skąd bierze się choroba
Źródłem problemu jest zwykle wariant w ABCD1, czyli genie odpowiedzialnym za prawidłowy transport tłuszczów do peroksysomów. Gdy ten mechanizm zawodzi, w organizmie gromadzą się bardzo długie kwasy tłuszczowe (VLCFA), które z czasem uszkadzają mielinę oraz korę nadnerczy. W praktyce oznacza to, że choroba dotyczy nie tylko „mózgu” albo tylko „hormonów”, lecz całego układu, który od tych struktur zależy.
Z tego powodu ALD nie zachowuje się jak jedna prosta jednostka chorobowa. Ten sam wariant genetyczny może dać inny przebieg u braci, kuzynów czy matek i córek w tej samej rodzinie. To ważne, bo intuicja często podpowiada, że wynik genetyczny powinien od razu „powiedzieć wszystko”, a przy ALD tak nie jest.
Jakie są główne postacie
| Postać | Typowy wiek lub płeć | Dominujące objawy | Najważniejszy cel leczenia |
|---|---|---|---|
| Mózgowa postać dziecięca | Najczęściej chłopcy w dzieciństwie | Zmiana zachowania, trudności w nauce, zaburzenia widzenia, spastyczność, napady | Wykrycie zmian na MRI i szybka kwalifikacja do leczenia ośrodkowego |
| Adrenomieloneuropatia | Najczęściej dorośli mężczyźni, czasem kobiety | Sztywność nóg, ból, trudność w chodzeniu, pęcherz i jelita, zaburzenia seksualne | Stała opieka neurologiczna i endokrynologiczna |
| Izolowana niewydolność nadnerczy | W każdym wieku | Osłabienie, chudnięcie, wymioty, przebarwienie skóry, spadki ciśnienia | Szybkie leczenie hormonalne i dalsze szukanie zajęcia układu nerwowego |
| Objawy u kobiet z wariantem ABCD1 | Zwykle dorosłość | Łagodniejsza, ale realna spastyczność i problemy z pęcherzem lub jelitami | Nie bagatelizować sygnałów i planować długoterminową kontrolę |
W ALD szczególnie ważny jest jeden niuans: przebieg nie wynika wprost z wyniku badania genetycznego ani z poziomu VLCFA. To oznacza, że przewidywanie choroby wyłącznie „po mutacji” jest zbyt uproszczone. Zapamiętaj: ten sam wariant ABCD1 może dać bardzo różny obraz kliniczny nawet w obrębie jednej rodziny.
Przeczytaj również: Choroby i schorzenia
Jakie objawy powinny wzbudzić czujność?
Najbardziej alarmuje połączenie kilku objawów: zmiana zachowania, spadek szkolnej formy, sztywność nóg albo cechy niewydolności nadnerczy. Jedna nieswoista dolegliwość nie przesądza o ALD, ale zestaw sygnałów już wymaga szybkiej oceny.
U dzieci
U chłopców pierwsze sygnały bywają zaskakująco „niemedyczne”. Pojawiają się kłopoty z koncentracją, impulsywność, pogorszenie wyników w nauce, gorsze rozumienie poleceń albo wycofanie społeczne. Dopiero później dochodzą zaburzenia chodu, sztywność, problemy z widzeniem, napady padaczkowe lub wyraźny regres rozwojowy. To właśnie ten etap jest najbardziej zdradliwy, bo łatwo wytłumaczyć go stresem, nadmiarem bodźców albo problemami szkolnymi.
Nie każdy spadek formy w szkole oznacza chorobę rzadką. Jeśli jednak obok trudności poznawczych pojawiają się objawy ruchowe albo wskazówki endokrynologiczne, próg podejrzenia powinien być niski. Uwaga: w ALD czas nie działa neutralnie, bo część zmian w mózgu rozwija się przed pełnym obrazem objawów neurologicznych.
U dorosłych
U dorosłych mężczyzn częściej dominuje adrenomieloneuropatia, czyli postać z wolnym, ale stałym narastaniem sztywności i osłabienia kończyn dolnych. Dochodzą trudności z szybkim chodzeniem, potykanie się, ból neuropatyczny, zaburzenia pęcherza, jelit i funkcji seksualnych. Taki obraz bywa mylony z chorobami kręgosłupa, zwyrodnieniem lub „niespecyficzną spastycznością”, przez co właściwa diagnoza opóźnia się o lata.
Warto zwrócić uwagę na sytuacje, w których objawy ruchowe łączą się z przewlekłym zmęczeniem, spadkami ciśnienia, ciemnieniem skóry albo nagłymi wymiotami. Wtedy trzeba myśleć nie tylko o neurologii, ale też o nadnerczach. W ALD te dwa obszary często biegną równolegle, choć nie zawsze w tym samym tempie.
U kobiet
U kobiet z wariantem ABCD1 problem bywa długo subtelny. Zwykle nie daje dziecięcych powikłań mózgowych, ale w dorosłości może pojawić się spastyczność, sztywność, ból i zaburzenia pracy pęcherza lub jelit. Największy błąd polega na traktowaniu ich wyłącznie jako „nosicielek”, bo objawy, choć często łagodniejsze niż u mężczyzn, są realne i wymagają oceny.
To szczególnie ważne w rodzinach, w których ALD już się pojawiło. Dodatni wywiad rodzinny zmienia wagę nawet drobnych objawów, bo pozwala szybciej powiązać neurologię z genetyką. Właśnie dlatego pytanie o krewnych nie jest dodatkiem do wywiadu, tylko jednym z głównych narzędzi rozpoznania.
Najbardziej pilne objawy to nagłe wymioty, osłabienie, omdlenie, narastająca senność, szybkie pogorszenie chodu, utrata mowy lub widzenia oraz zachowania sugerujące regres poznawczy. Taki zestaw wymaga szybkiej oceny lekarskiej, bo może oznaczać zarówno zajęcie mózgu, jak i niewydolność nadnerczy.
Zapamiętaj: ALD u dzieci nie zaczyna się zawsze od „klasycznej” neurologii. Czasem pierwszym sygnałem są szkolne trudności albo niespecyficzne osłabienie.
Przeczytaj również: Choroby rzadkie - Jak rozpoznać objawy i gdzie szukać pomocy?
Jak rozpoznaje się ALD i które badania są kluczowe?
Rozpoznanie opiera się na badaniu VLCFA, analizie genu ABCD1, ocenie hormonów nadnerczy i MRI mózgu. Sam obraz kliniczny nie wystarcza, bo fenotyp ALD bywa zmienny nawet w obrębie jednej rodziny, a objawy potrafią wyprzedzać lub opóźniać zmiany laboratoryjne.
Według GeneReviews podejrzenie ALD pojawia się najczęściej w trzech sytuacjach: po dodatnim przesiewie noworodkowym, u osoby już objawowej albo u chłopca wykrytego w badaniu rodzinnym. To ważne, bo wczesne wykrycie pozwala rozpocząć nadzór zanim wystąpią nieodwracalne uszkodzenia.
Badania biochemiczne i genetyczne
Podstawą są bardzo długie kwasy tłuszczowe w surowicy oraz potwierdzenie wariantu w ABCD1. U mężczyzn badanie biochemiczne zwykle jest bardzo czytelne, natomiast u kobiet wynik może być mniej jednoznaczny, dlatego analiza genetyczna zyskuje wtedy jeszcze większe znaczenie. W praktyce liczy się nie jeden wynik, lecz zgodność danych klinicznych, rodzinnych i laboratoryjnych.
Warto też pamiętać, że sam dodatni wynik badania przesiewowego nie oznacza pełnego rozpoznania. Jest sygnałem do pilnego potwierdzenia choroby i uruchomienia monitorowania. Uwaga: u chłopców z dodatnim przesiewem czas do pierwszej oceny endokrynologicznej i neurologicznej ma bezpośredni wpływ na bezpieczeństwo.
Obserwacja nadnerczy i mózgu
| Wiek lub sytuacja | Kontrola ACTH i kortyzolu | MRI mózgu | Co to daje |
|---|---|---|---|
| Po dodatnim wyniku przesiewu u chłopca | Natychmiastowa ocena endokrynologiczna | Plan tworzony od razu po potwierdzeniu rozpoznania | Wczesne wychwycenie niewydolności nadnerczy i zmian w ośrodkowym układzie nerwowym |
| Od 12. miesiąca do 3. roku życia | Co 3–6 miesięcy aż do 10. roku życia, potem rzadziej | Raz w roku | Wykrycie zmian zanim dziecko zacznie gorzej funkcjonować |
| Od 3. do 12. roku życia | Co 3–6 miesięcy aż do 10. roku życia, potem zwykle raz w roku | Co 6 miesięcy | Najwyższe ryzyko wejścia w postać mózgową wymaga częstszego nadzoru |
| Powyżej 12. roku życia | Zwykle co najmniej raz w roku | Raz w roku | Cerebralne zmiany mogą pojawić się także w dorosłości |
Taki harmonogram może wyglądać na nadmiarowy, ale właśnie on zmienia rokowanie. Zmiany na MRI potrafią pojawić się wcześniej niż wyraźne objawy neurologiczne, a drobne odchylenia w hormonach nadnerczy wyprzedzają pełnoobjawową niewydolność. Dzięki temu decyzje terapeutyczne można podejmować wtedy, gdy organizm ma jeszcze większą szansę na ochronę.
Kiedy potrzebne jest pilne działanie
Najbardziej pilna sytuacja to chłopiec lub mężczyzna z objawami niewydolności nadnerczy, zwłaszcza jeśli pojawia się osłabienie, wymioty, hipotensja albo zaburzenia świadomości. Drugim alarmem są nowe zmiany w zachowaniu, nauce lub chodzie, szczególnie gdy obraz rozwija się szybko. Wtedy nie czeka się na „pełny” zestaw objawów, tylko uruchamia diagnostykę i konsultację w ośrodku znającym ALD.
W krajach, które prowadzą przesiew noworodków w kierunku ALD, pierwsze decyzje zapadają właśnie na tym etapie. To pokazuje, że w tej chorobie prawdziwą przewagę daje nie dramatyczne rozpoznanie, lecz wcześniejsze uchwycenie subtelnego sygnału. Największy błąd diagnostyczny polega na czekaniu, aż obraz stanie się oczywisty.
Uwaga: Negatywny opis MRI nie zamyka tematu. U chłopców z potwierdzonym ALD nadzór trwa dalej, bo ryzyko zmiany obrazu pozostaje realne.
Jak wygląda leczenie i co działa najlepiej?
Najlepsze wyniki daje leczenie wdrożone wcześnie, zanim pojawi się zaawansowane uszkodzenie mózgu. W ALD nie ma jednego uniwersalnego schematu, bo terapia zależy od tego, która postać dominuje i na jakim etapie choroba została wykryta.
Według międzynarodowego konsensusu opublikowanego w Neurology najważniejsze są: wczesne rozpoznanie, regularny nadzór, leczenie endokrynologiczne i kwalifikacja do terapii ośrodkowej wtedy, gdy zmiany mózgowe są jeszcze małe. To podejście nie brzmi spektakularnie, ale właśnie ono ratuje funkcję, którą łatwo stracić bezpowrotnie.
Nadnercza
Jeżeli dochodzi do niewydolności kory nadnerczy, leczenie polega na substytucji hormonalnej prowadzonej przez endokrynologa. Nie chodzi tu o „wspomaganie”, ale o zastąpienie brakujących hormonów w sposób, który chroni przed przełomem nadnerczowym. To jeden z tych elementów, w których szybka reakcja ma bezpośrednie znaczenie dla życia.
Równolegle trzeba kontrolować, czy nie pojawiają się objawy neurologiczne. U wielu osób przerwanie obserwacji po samym ustawieniu hormonów byłoby błędem, bo ALD nie zatrzymuje się na jednym układzie. W praktyce: opieka endokrynologiczna i neurologiczna powinny iść razem, a nie po kolei.
Mózgowa postać
W mózgowej postaci dziecięcej najważniejsze są HSCT, czyli przeszczepienie krwiotwórczych komórek macierzystych, oraz w wybranych ośrodkach terapia genowa ex vivo. Największą korzyść daje leczenie u chłopców bez ciężkich objawów neurologicznych, ale już z charakterystycznymi zmianami w MRI. Gdy deficyty są rozległe, korzyść z takiej interwencji wyraźnie maleje, dlatego liczy się moment kwalifikacji.
To także obszar, w którym najłatwiej przeoczyć szansę. Chłopiec może jeszcze chodzić, mówić i funkcjonować w domu, a mimo to w MRI już pojawia się początek procesu zapalno-demielinizacyjnego. Dlatego samo „brak wyraźnych objawów” nie wystarcza jako uspokojenie, jeśli badanie obrazowe pokazuje niepokojące zmiany.
Wsparcie długoterminowe
Poza leczeniem przyczynowym potrzebne są rehabilitacja, logopedia, wsparcie psychologiczne, kontrola pęcherza i jelit oraz planowanie opieki rodzinnej. U części osób problemem staje się też jedzenie, mowa, mobilność i bezpieczeństwo w domu. ALD wymaga więc nie tylko terapii jednego narządu, ale także organizacji życia wokół zmieniających się potrzeb chorego.
Ważną częścią opieki pozostaje też poradnictwo genetyczne. Dzięki niemu można zidentyfikować krewnych męskich, którzy jeszcze nie mają objawów, ale już wymagają nadzoru. To jeden z niewielu obszarów medycyny, w których dobrze poprowadzona rodzina zyskuje realną przewagę nad chorobą.
W praktyce: Dobre leczenie ALD zaczyna się dużo wcześniej niż od leku. Zaczyna się od ośrodka, który zna harmonogram kontroli, wie kiedy kierować na MRI i nie bagatelizuje hormonów nadnerczy.
Przeczytaj również: Zwłóknienie płuc - Jak rozpoznać objawy i spowolnić postęp choroby?
Co oznacza ALD w Polsce?
W Polsce ALD figuruje jako choroba rzadka z kodem ORPHA 43, ale nie ma jej w standardowym panelu przesiewu noworodków. To oznacza, że droga do diagnozy częściej zaczyna się od czujności lekarza, rodziny albo pierwszych nietypowych objawów niż od rutynowego programu populacyjnego.
Na stronie Ministerstwa Zdrowia widnieje obecny program badań przesiewowych noworodków, który obejmuje m.in. hipotyreozę, PKU, mukowiscydozę, rzadkie wady metabolizmu, wrodzony przerost nadnerczy, deficyt biotynidazy i SMA. W tej liście ALD nie figuruje, więc rodziny obciążone wywiadem nie mogą liczyć wyłącznie na standardowy przesiew.
Jak wygląda przesiew
Ograniczeniem polskiego systemu nie jest brak wiedzy o ALD, tylko brak powszechnego włączenia tej choroby do standardowego panelu. Dlatego każde dziecko z niepokojącymi objawami, a zwłaszcza z dodatnim wywiadem rodzinnym, powinno trafić do neurologa, endokrynologa i genetyka. W takiej sytuacji rodzinne obciążenie jest tak samo ważne jak aktualny wynik badania.
W materiałach parlamentarnych przywoływano częstość 1:16800 żywych urodzeń: Sejm. To dobry przykład tego, jak rzadka choroba potrafi pozostać poza codzienną czujnością systemu, choć jej konsekwencje są bardzo poważne.
Gdzie szukać pomocy
Najkrótsza ścieżka prowadzi przez lekarza rodzinnego lub pediatrę do specjalisty, a potem do ośrodka z doświadczeniem w chorobach rzadkich. Pomocne są też publiczne bazy i poradniki dla chorób rzadkich, bo pozwalają szybciej ustalić, gdzie w kraju dostępne są badania, poradnictwo i opieka wielospecjalistyczna. W ALD nie chodzi o „jednego lekarza od wszystkiego”, tylko o zespół, który potrafi zsynchronizować neurologię, hormony i genetykę.
W praktyce warto pamiętać o jednej zasadzie: jeżeli w rodzinie rozpoznano ALD, nie czeka się biernie na objawy u innych krewnych. Badanie mężczyzn z rodziny, a w odpowiednich sytuacjach także konsultacja kobiet, pozwala wychwycić ryzyko zanim pojawi się niewydolność nadnerczy albo postać mózgowa.
Najczęstsze błędy i ścieżka działania
- Traktowanie dziewczynki z wariantem ABCD1 jak osoby bez problemu - objawy mogą pojawić się później i wymagają obserwacji.
- Czekanie na pełnoobjawowy obraz - w ALD to zwykle opóźnia leczenie zamiast je ułatwiać.
- Ograniczenie diagnostyki do jednego badania - potrzebne są dane kliniczne, biochemiczne, genetyczne i obrazowe.
- Brak planu nadzoru - bez harmonogramu ACTH, kortyzolu i MRI łatwo przegapić moment kwalifikacji do leczenia.
Najbezpieczniejsza ścieżka wygląda prosto: szybkie podejrzenie, potwierdzenie laboratoryjne, ocena hormonalna, MRI, konsultacja genetyczna i decyzja w ośrodku referencyjnym. Ta kolejność jest ważniejsza niż próba samodzielnego dopasowania objawów do internetowych opisów. Jeżeli ALD wchodzi w grę, czas liczy się bardziej niż intuicja.
W praktyce: Najwięcej traci się wtedy, gdy czeka się na „pełen” obraz choroby. ALD trzeba łapać na etapie subtelnych zmian.
Największą różnicę robi szybkie połączenie neurologii, endokrynologii i genetyki, bo właśnie wtedy można jeszcze zatrzymać część najcięższych powikłań.